Simulation
This simulation completely generates genotypes (G) and phenotypes (Y) for multiple simulated ancestries while controlling for the heritability (
flowchart TB
geneInput --> common
input --> SNPeffects
subpopulations --> subjGeno
subjGeno --> geneEffects
ethBalance --> subjAncestry
siteInput --> balance
causalShared -->geneEffects
balance --> subjSite
geneInput --> causalShared
subgraph User Input
geneInput("# SNPs, (M) \n # Ancestries (O) \n Proportion causal (p) \n Proportion similar, p(Shared)")
siteInput("# subjects (N) \n # sites (N_S) \n balance")
input("Heritability, h2")
end
subgraph Allele frequencies
causalShared(Simulate Causal and shared regions) --> common
common(Simulate \n Common ancestry) --> subpopulations(Simulate \n modern ancestries)
end
subgraph Study design
balance(Simulate numbers \n at each site) --> ethBalance(Simulate ancestries \n at each site)
end
subgraph Subjects
subjSite(Assign subject \n Site)
subjAncestry(Sample subject \n Ancestry)
subjGeno(Sample subject \n genotype)
subjSite --> subjAncestry --> subjGeno
end
subgraph Effects
SNPeffects(Simulate SNP effects)
geneEffects(Compute gene \n effects)
SiteEffects(Simulate Site effects)
errors(Simulate \n noise)
phenotype(Simulate phenotype)
SNPeffects --> geneEffects
geneEffects --> phenotype
SiteEffects --> phenotype
errors --> phenotype
end
Simulations can be run off of provided plink formatted (.bed, .bim, .fam) genotypes. If the sample is known to come from mixed ancestries, an additional subject ancestry file can be provided which should have the following format
IID, FID, subj_ancestries
a, 1, AFR
b, 2, EUR
...
Note this is a comma separated file and the column names need to be precisely as defined above. The values for subj_ancestries can take any string and so doesn't need to be 3 capital letters.
The simulation starts by simulating Single Nucleotide Polymorphisms at
- Simulating allelic frequency of a "common ancester"
- Simulating allelic frequencies of a set of ancestries that are descendent from the "common ancester"
- Simulate subject genotypes based on their assigned ancestry
Each of these steps are outlined in further detail below
("Simulate/simulation_helpers/sites.py" sim_sites)
The study design refers to the number of subjects total, the number of subjects of each reported ancestry, and, if desired, the distribution of those subjects across multiple sites.
Simulation of imbalanced study designs are possible both in terms of subjects at each site (
Note that right now this is restricted to a symmetric dirichlet parameter (i.e.
("Simulate/simulation_helpers/clusters.py" assign_clusters)
Ancestries can be either assigned equally across all sites, their overall proportions sampled from a symmetric dirichlet distribution in which case the numbers of each ancestry may differ across each site.
Differentiation between ancestries is achieved by changing the allele frequencies (
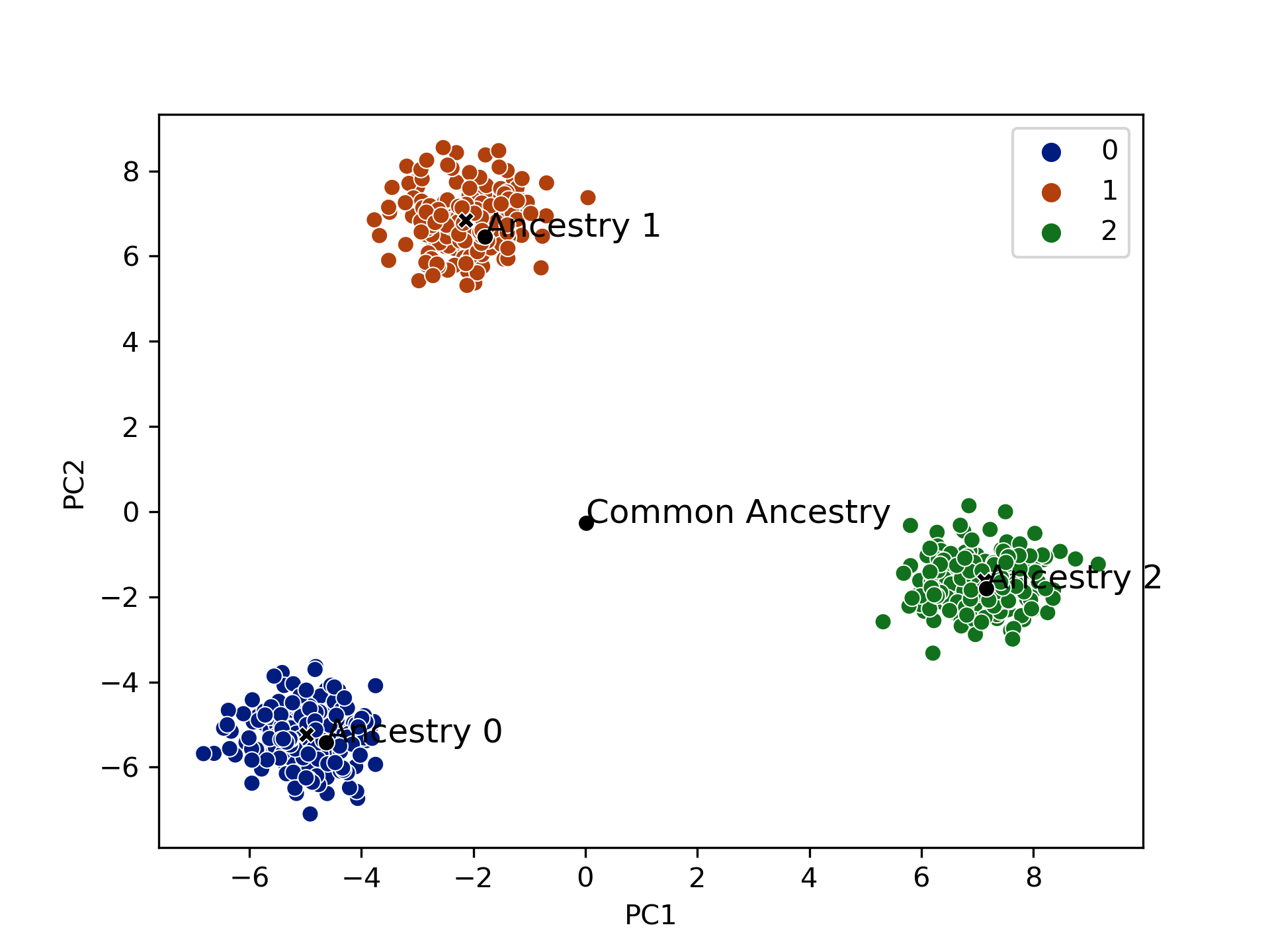
%%| label: fig-ancestries
%%| caption: Diagram of deriving ancestries from a common ancestor.
flowchart TB
common(Common Ancestry, f_*0)
a1(Ancestry 1, f_*1)
a2(Ancestry 2, f_*2)
more("...")
aO(Ancestry O, f_*O)
common --> a1
common -->a2
common --> more
common --> aO
The common ancester is defined by a vector of allelic frequencies (
$$ ∀ m∈ {1, …, M}, f_{m0} \stackrel{iid}{\sim} Uni(0.1, 0.9) \ f_{*0} \sim Unif(0.1, 0.9)\ $$ ("Simulate/simulation_helpers/clusters.py")
Furthermore, for simulation purposes, we assume that the resulting modern ancestries derived from said common ancestor are derived from stochastic differences in allelic frequencies. More precisely, we assume that each ancestry is a random realization of a Beta distribution with the same mean as the common ancestry and an appropriately chosen variance. This amounts to a sampling as follows
This supoposes the allele frequencies of the newer ancestors are random flucuations centered at the allele frequencies of the common ancestor. This is because the mean for the
And the variance is
Therefore, the simulated allele frequencies will be centered at allele frequencies of the common ancestor, and the spread of the resulting ancestries will be random fluctions around that center controlled by
("Simulate/simulation_helpers/genos.py" sim_genos)
The genotypes of each of the subjects are assumed to be independent draws from a
("Simulate/simulation_helpers/pheno.py" sim_pheno)
The effects are simulated such that the phenotypic variance attributable to SNP effects (as opposed to random variances), also defined as the heritabilty (
If there it is assumed that there are effects shared between ancestries (homogeneous) and effects that are specific to ancestries, the heritability for each ancestry (
While, this doesn't fix the heritability for ancestry o precisely to the specified heritability it is generally very close.
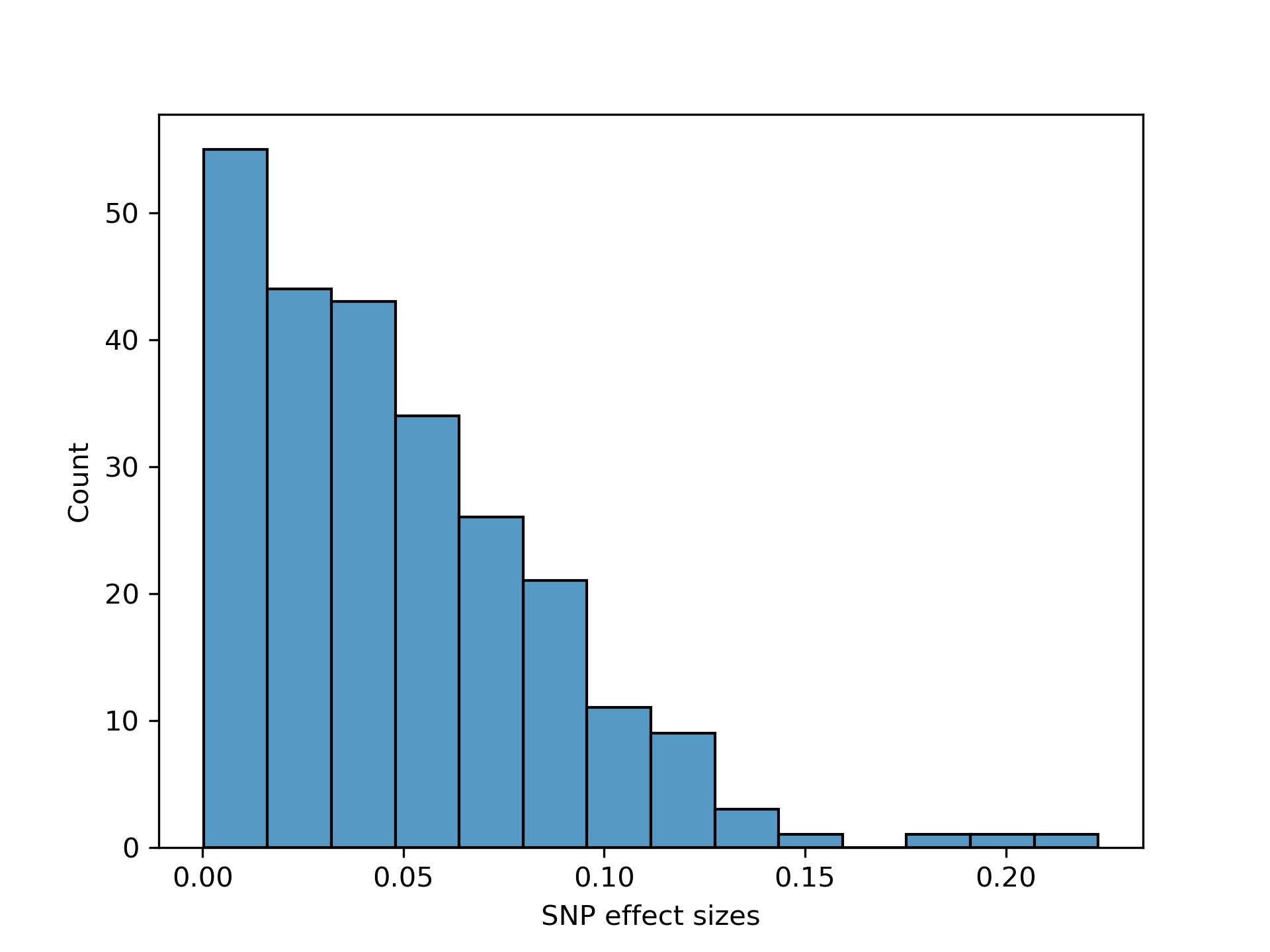
The SNPs effects are supposed to be inversely related to their respective minor allelic frequencies. So, to this end, the SNP effects are sampled as follows
Some portion of the effects are assumed to be homogeneous (w.r.t. ancestry) and so the allele frequency is taken to be the overall allele frequency in the overall sample. Some portion of the effects are assumed to be heterogeneous (w.r.t. ancestry) and so the allele frequency is taken to be the allele frequency within the given ancestry.
The random noise is simulated so as to satisfy the prescribed heritability so
and in the presence of hetergenous effects
The site effects aren't assumed to be correlated with the SNP effects and so can either be sample from a normal distribution as
The phenotypes are simulated from a linear model combining all of the above effects as
- The heritability is controlled to a prespecified value (
$h^2$ ) - The affect of a given SNP is related to it's allelic frequency
It has been suggested that a SNP effect is related to its observed frequency. ADD A CITATION AND AN IMAGE FOR THAT.
| Symbol | Definition |
|---|---|
| Number of sites | |
| Number of SNPs | |
| Number of subjects | |
| Number of ancestries | |
| site index |
|
| SNP index, |
|
| Subject index, |
|
| Ancestry index index, |
|
| allelic frequencies, dimension |
|
| column vector of allelic frequencies for ancestry |
|
| variance associated with simulating allelic frequencies of the ancestries derived from the common ancestor | |
| Proportion of genome considered causal (with nonzero effect) | |
|
|
|
| is number of subjects at site l |